Optic Neuritis: Understanding Vision Loss, Recovery, and Hope for the Future
Optic neuritis explained: symptoms, treatment, recovery, and hope. Learn how to support someone with vision loss and explore future treatments like bionic eyes.
DISABLED ENTREPRENEUR – DISABILITY UK
Disability UK Online Health Journal – All In One Business In A Box – Forum – Business Directory – Useful Resources – Health – Human Rights – Politics
DISABLED ENTREPRENEUR – DISABILITY UK
Disability UK Online Health Journal – All In One Business In A Box – Forum – Business Directory – Useful Resources – Health – Human Rights – Politics
Browsing Category
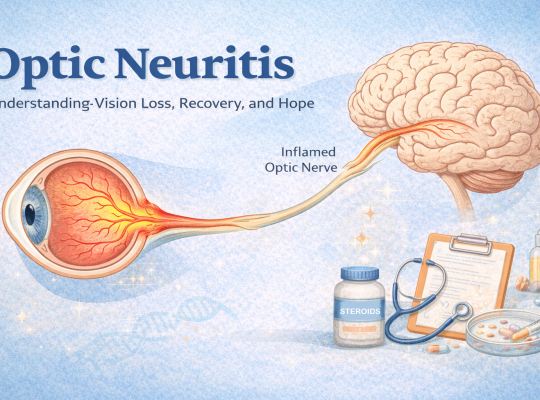
Optic neuritis explained: symptoms, treatment, recovery, and hope. Learn how to support someone with vision loss and explore future treatments like bionic eyes.

Every new discovery in genetics brings scientists one step closer to understanding complex conditions and improving the lives of those affected. Learn how MIRAGE syndrome was discovered in 2016 through genetic sequencing and why this rare SAMD9-related disorder is one of the rarest medical conditions in the world.

Multiple sclerosis is a complex and challenging condition to manage, but a wide range of therapies offers hope for patients. From disease-modifying drugs like Alemtuzumab and Anti-CD20 therapies to natural approaches such as sun therapy, each treatment targets different aspects of the disease.

True wellness isn’t about perfecting one area of your health while neglecting others. It’s about recognizing that mental clarity, physical vitality, and nutritional choices are deeply intertwined, each one influencing the others in ways that can either elevate or undermine your overall well-being.

“China unveils the world’s first pregnancy robot with an artificial womb. A breakthrough, or are we playing God? Explore the ethics, risks, and future of birth.”

Disclaimer: This article is for informational purposes only and should not be considered a substitute for professional medical advice, diagnosis, or treatment. Always seek advice from qualified healthcare professionals regarding …

To stay on top in the advertising world, think about using AI-powered AdTech in your marketing plans. When you adopt these new ideas, you can boost how well your ads work, fine-tune your campaigns, and build strong ties with your audience. Get ahead of other companies by putting money into AI tech now, and open up new chances to grow and do well in tomorrow’s advertising landscape.

Mapping the DNA of every baby born in the UK is a bold and questionable futuristic leap in public health. If implemented with the right ethical safeguards and public engagement, it could set a global standard for proactive healthcare, reduce disease burden, and improve life expectancy across generations. But as science marches forward, so must human rights protections. The challenge is not just mapping genes, it’s mapping out a future where technology enhances freedom, dignity, and equality, not erodes them.

In a historic medical breakthrough, a three-year-old child has become the youngest patient ever to receive a revolutionary form of gene therapy, offering hope to families affected by rare and life-threatening genetic conditions. The pioneering treatment, administered at a leading children’s hospital in the UK, has shown early signs of success and could change the trajectory of treatment for similar disorders worldwide.
A groundbreaking and highly controversial scientific initiative is now underway in the UK, as leading British researchers aim to synthesize the first human genome entirely from scratch, not by altering existing DNA, but by building it letter by letter in the lab. Spearheaded by scientists from the University of Oxford, Cambridge, Kent, Manchester, and Imperial College London, this ambitious project is known as the Synthetic Human Genome (SynHG) project and is being funded by the Wellcome Trust, the world’s largest medical research charity.